NEWTON, Mass.,
|
|||||
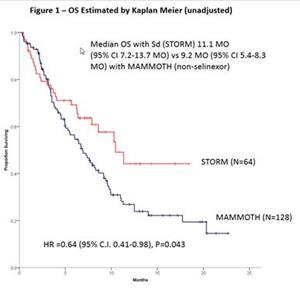
A key abstract at this meeting is a poster presentation titled “Outcomes of Triple Class Refractory Penta-Exposed Multiple Myeloma (MM),” (Cornell, et al) which compares the overall survival (OS) rate from the retrospective MAMMOTH study (Leukemia, 2019) which evaluated outcomes from patients with relapsed or refractory multiple myeloma after their disease became refractory to CD38 monoclonal antibodies with a similarly-matched cohort of patients from Karyopharm’s Phase 2b STORM study. Patients in STORM, who received selinexor and dexamethasone as first line of therapy after their disease became triple class refractory (n=64) as compared with matched patients receiving currently available therapies from the MAMMOTH cohort (n=128) showed an unadjusted hazard ratio (HR) for death of 0.64 (p=0.043), while an adjusted analysis, which takes into consideration differences in baseline characteristics between the two groups, showed a HR of 0.55 (p=0.009) (see Figure 1 below). The median OS on selinexor-dexamethasone was 11.1 months and on MAMMOTH was 9.2 months. Patients in the MAMMOTH study received a single (n=6, 4.7%) or combination of two or more anti-multiple myeloma agents (n=122, 95.3%). A PDF copy of this poster will be available here once it has been presented at the meeting.
Additional abstracts to be presented include an oral presentation highlighting updated data from the Phase 1b/2 STOMP study (White, et al) evaluating selinexor and dexamethasone in combination with Revlimid® (lenalidomide) for the treatment of patients with relapsed or refractory multiple myeloma, and three additional posters (Gavriatopoulou et al, Tariq et al and Vogl et al) which describe various analyses from the Phase 2b STORM study evaluating XPOVIO in patients with heavily pretreated multiple myeloma.
“The multiple data presentations at IMW continue to reinforce our belief in the potential of XPOVIO as a new therapeutic option for patients with relapsed or refractory multiple myeloma,” said Sharon Shacham, PhD, MBA, President and Chief Scientific Officer of Karyopharm. “There is a growing need for novel treatment approaches for patients with relapsed or refractory multiple myeloma. The unique mechanism of action of XPOVIO, the first and only oral nuclear export inhibitor approved in the U.S., provides physicians with a new therapeutic option for patients with heavily pretreated multiple myeloma and we continue to be excited about future development of this novel agent.”
Details for the IMW 2019 presentations are as follows:
Oral Presentation
Title: Safety and Efficacy of the Combination of Selinexor, Lenalidomide and Dexamethasone (SRd) in Patients with Relapsed/Refractory Multiple Myeloma (RRMM)
Lead author: Darrell White,
Abstract #: AB353
Session: Multiple Myeloma Novel Agents
Date and Time:
Location:
Poster Presentations
Title: Outcomes of Triple Class Refractory Penta-Exposed Multiple Myeloma (MM)
Lead author: Robert Cornell,
Abstract #: 866
Session: Multiple Myeloma Novel Agents – Poster Session I
Date and Time:
Location:
Title: Effect of Age on the Safety and Efficacy of Selinexor in Patients with Relapsed Refractory Multiple Myeloma: A Post-hoc Analysis of the STORM Study
Lead author: Maria Gavriatopoulou,
Abstract #: 582
Session: Multiple Myeloma Novel Agents – Poster Session I
Date and Time:
Location:
Title: Efficacy and Safety of Selinexor for Heavily Pretreated Multiple Myeloma Treatment – A Systematic Review
Lead author: Muhammad Junaid Tariq, John H. Stroger,
Abstract #: 617
Session: Multiple Myeloma Novel Agents – Poster Session I
Date and Time:
Location:
Title: Improvements in Renal Function with Selinexor in Relapsed/Refractory Multiple Myeloma: Post-hoc Analyses from the STORM Study
Lead author: Dan Vogl,
Abstract #: 592
Session: Multiple Myeloma Novel Agents – Poster Session I
Date and Time:
Location:
About XPOVIO™ (selinexor)
XPOVIO is a first-in-class, oral Selective Inhibitor of Nuclear Export (SINE) compound. XPOVIO functions by selectively binding to and inhibiting the nuclear export protein exportin 1 (XPO1, also called CRM1). XPOVIO blocks the nuclear export of tumor suppressor, growth regulatory and anti-inflammatory proteins, leading to accumulation of these proteins in the nucleus and enhancing their anti-cancer activity in the cell. The forced nuclear retention of these proteins can counteract a multitude of the oncogenic pathways that, unchecked, allow cancer cells with severe DNA damage to continue to grow and divide in an unrestrained fashion. In addition to receiving accelerated FDA approval of XPOVIO in
IMPORTANT SAFETY INFORMATION
Thrombocytopenia
XPOVIO can cause thrombocytopenia, leading to potentially fatal hemorrhage. Thrombocytopenia was reported as an adverse reaction in 74% of patients, and severe (Grade 3-4) thrombocytopenia occurred in 61% of patients treated with XPOVIO. The median time to onset of the first event was 22 days. Bleeding occurred in 23% of patients with thrombocytopenia, clinically significant bleeding occurred in 5% of patients with thrombocytopenia and fatal hemorrhage occurred in <1% of patients.
Monitor platelet counts at baseline, during treatment, and as clinically indicated. Monitor more frequently during the first two months of treatment. Institute platelet transfusion and/or other treatments as clinically indicated. Monitor patients for signs and symptoms of bleeding and evaluate promptly. Interrupt and/or reduce dose, or permanently discontinue based on severity of adverse reaction.
Neutropenia
XPOVIO can cause neutropenia, potentially increasing the risk of infection. Neutropenia was reported as an adverse reaction in 34% of patients, and severe (Grade 3-4) neutropenia occurred in 21% of patients treated with XPOVIO. The median time to onset of the first event was 25 days. Febrile neutropenia was reported in 3% of patients.
Obtain neutrophil counts at baseline, during treatment, and as clinically indicated. Monitor more frequently during the first two months of treatment. Monitor patients for signs and symptoms of concomitant infection and evaluate promptly. Consider supportive measures including antimicrobials for signs of infection and use of growth factors (e.g., G-CSF). Interrupt and/or reduce dose, or permanently discontinue based on severity of adverse reaction.
Gastrointestinal Toxicity
Gastrointestinal toxicities occurred in patients treated with XPOVIO.
Nausea/Vomiting
Nausea was reported as an adverse reaction in 72% of patients, and Grade 3 nausea occurred in 9% of patients treated with XPOVIO. The median time to onset of the first nausea event was 3 days.
Vomiting was reported in 41% of patients, and Grade 3 vomiting occurred in 4% of patients treated with XPOVIO. The median time to onset of the first vomiting event was 5 days.
Provide prophylactic 5-HT3 antagonists and/or other anti-nausea agents, prior to and during treatment with XPOVIO. Manage nausea/vomiting by dose interruption, reduction, and/or discontinuation. Administer intravenous fluids and replace electrolytes to prevent dehydration in patients at risk. Use additional anti-nausea medications as clinically indicated.
Diarrhea
Diarrhea was reported as an adverse reaction in 44% of patients, and Grade 3 diarrhea occurred in 6% of patients treated with XPOVIO. The median time to onset of diarrhea was 15 days.
Manage diarrhea by dose modifications and/or standard anti-diarrheal agents; administer intravenous fluids to prevent dehydration in patients at risk.
Anorexia/Weight Loss
Anorexia was reported as an adverse reaction in 53% of patients, and Grade 3 anorexia occurred in 5% of patients treated with XPOVIO. The median time to onset of anorexia was 8 days.
Weight loss was reported as an adverse reaction in 47% of patients, and Grade 3 weight loss occurred in 1% of patients treated with XPOVIO. The median time to onset of weight loss was 15 days.
Monitor patient weight at baseline, during treatment, and as clinically indicated. Monitor more frequently during the first two months of treatment. Manage anorexia and weight loss with dose modifications, appetite stimulants, and nutritional support.
Hyponatremia
XPOVIO can cause hyponatremia; 39% of patients treated with XPOVIO experienced hyponatremia, 22% of patients experienced Grade 3 or 4 hyponatremia. The median time to onset of the first event was 8 days.
Monitor sodium level at baseline, during treatment, and as clinically indicated. Monitor more frequently during the first two months of treatment. Correct sodium levels for concurrent hyperglycemia (serum glucose >150 mg/dL) and high serum paraprotein levels. Treat hyponatremia per clinical guidelines (intravenous saline and/or salt tablets), including dietary review. Interrupt and/or reduce dose, or permanently discontinue based on severity of adverse reaction.
Infections
In patients receiving XPOVIO, 52% of patients experienced any grade of infection. Upper respiratory tract infection of any grade occurred in 21%, pneumonia in 13%, and sepsis in 6% of patients. Grade ≥3 infections were reported in 25% of patients, and deaths resulting from an infection occurred in 4% of patients. The most commonly reported Grade ≥3 infections were pneumonia in 9% of patients, followed by sepsis in 6%. The median time to onset was 54 days for pneumonia and 42 days for sepsis. Most infections were not associated with neutropenia and were caused by non-opportunistic organisms.
Neurological Toxicity
Neurological toxicities occurred in patients treated with XPOVIO.
Neurological adverse reactions including dizziness, syncope, depressed level of consciousness, and mental status changes (including delirium and confusional state) occurred in 30% of patients, and severe events (Grade 3-4) occurred in 9% of patients treated with XPOVIO. Median time to the first event was 15 days.
Optimize hydration status, hemoglobin level, and concomitant medications to avoid exacerbating dizziness or mental status changes.
Embryo-Fetal Toxicity
Based on data from animal studies and its mechanism of action, XPOVIO can cause fetal harm when administered to a pregnant woman. Selinexor administration to pregnant animals during organogenesis resulted in structural abnormalities and alterations to growth at exposures below those occurring clinically at the recommended dose.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential and males with a female partner of reproductive potential to use effective contraception during treatment with XPOVIO and for 1 week after the last dose.
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥20%) are thrombocytopenia, fatigue, nausea, anemia, decreased appetite, decreased weight, diarrhea, vomiting, hyponatremia, neutropenia, leukopenia, constipation, dyspnea, and upper respiratory tract infection.
The treatment discontinuation rate due to adverse reactions was 27%; 53% of patients had a reduction in the XPOVIO dose, and 65.3% had the dose of XPOVIO interrupted. The most frequent adverse reactions requiring permanent discontinuation in 4% or greater of patients who received XPOVIO included fatigue, nausea, and thrombocytopenia. The rate of fatal adverse reactions was 8.9%.
Please see XPOVIO Full Prescribing Information available at www.XPOVIO.com.
About Karyopharm Therapeutics
Forward-Looking Statements
This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995. Such forward-looking statements include those regarding our expectations relating to XPOVIO for the treatment of patients with RRMM, commercialization of XPOVIO or any of our drug candidates, submissions to, and the review and potential approval of selinexor by, regulatory authorities, including the anticipated timing of such submissions and actions and the potential availability of accelerated approval pathways, and the therapeutic potential of and potential clinical development plans for Karyopharm's drug candidates, especially selinexor. Such statements are subject to numerous important factors, risks and uncertainties, many of which are beyond Karyopharm's control, that may cause actual events or results to differ materially from Karyopharm's current expectations. For example, there can be no guarantee that regulators will agree that selinexor qualifies for conditional approval in the E.U. as a result of the data from the STORM study or accelerated or conditional approval in the U.S. or EU, respectively, based on data from the SADAL study in patients with relapsed or refractory DLBCL, or that any of Karyopharm's drug candidates, including selinexor, will successfully complete necessary clinical development phases or that development of any of Karyopharm's drug candidates will continue. Further, there can be no guarantee that any positive developments in Karyopharm's drug candidate portfolio will result in stock price appreciation. Management's expectations and, therefore, any forward-looking statements in this press release could also be affected by risks and uncertainties relating to a number of other factors, including the following: the timing and costs involved in commercializing XPOVIO or any of Karyopharm’s drug candidates that receive regulatory approval; the ability to retain regulatory approval of XPOVIO or any of Karyopharm’s drug candidates that receive regulatory approval; Karyopharm's results of clinical trials and preclinical studies, including subsequent analysis of existing data and new data received from ongoing and future studies; the content and timing of decisions made by the
Velcade® is a registered trademark of
Contacts:
Investors:
Ian Karp, Vice President, Investor and Public Relations
857-297-2241 | ikarp@karyopharm.com
Media:
Simona Kormanikova or Robert Stanislaro
212-850-5600 | Simona.Kormanikova@fticonsulting.com or robert.stanislaro@fticonsulting.com
A photo accompanying this announcement is available at https://www.globenewswire.com/NewsRoom/AttachmentNg/0becf145-4fcd-492e-94cd-52900a268433
![]()
Source: Karyopharm Therapeutics Inc.